Novel technologies in the life sciences produce information at an accelerating rate, with public data stores (such as the one managed by the European Bioinformatics Institute http://www.ebi.ac.uk) containing on the order of 10PB of biological information. For nearly 40 years, the same was not so for chemical information, but in 2004 a large public small-molecule structure repository (PubChem http://pubchem.ncbi.nlm.nih.gov) was made freely available by the National Library of Medicine (part of the U.S. National Institutes of Health) and soon followed by other databases. Likewise, while many of the foundational algorithms of cheminformatics have been described since the 1950s, open-source software implementing many of them have become accessible only since the mid-1990s.10
Key Insights
- Molecules with similar physical structure tend to have similar chemical properties.
- Open-source chemistry programs and open-access molecular databases allow interdisciplinary research opportunities that did not previously exist.
- Cheminformatics combines biological, chemical, pharmaceutical, and drug patient information to address large-scale data mining, curation, and visualization challenges.
Why is chemical information important? Why should chemists and computer scientists care about its public availability? And how does chemical information relate to the field of computer science? Though cheminformatics is used in fields from agrochemical research to the design of novel materials, here we use drug discovery as our context due to its relevance to human well-being. The art and science of drug discovery focuses on small molecules, with a core component of methods involving techniques and algorithms to handle, analyze, and interpret chemical-structure information. Unfortunately, drug-discovery research is slow, expensive, and prone to failure. Public availability of chemical information and related tools is important, as the more information available to each researcher, the better the chances of avoiding the many causes of attrition. Computer-science research is highly relevant to managing the volume and complexity of chemical information. A single database (such as PubChem) contains more than 34 million chemical-structure records, along with an even larger number of annotations (such as synonyms, or different names for the same molecule, with PubChem storing 50.1 million synonyms for 19.6 million compounds), known targets (drugs with which a molecule is known to interact), mode of action (how the molecule interacts with its targets), and status within the regulatory-approval process.
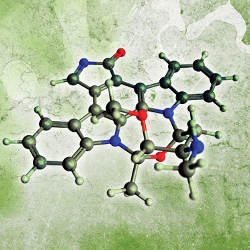
While bioinformatics often deals with sequences, the domain of cheminformatics is chemical structures. In the former, information is frequently represented as 1D strings that are relatively easy to handle computationally. In the latter, chemical structures are complex graphs that may include rings and branches, along with multiple valid representations of the same molecule to be considered for particular algorithms (such as tautomers, where hydrogen atoms are positioned at different places in the molecule). Hence, chemical structures are more difficult to standardize, and algorithms dealing with them must take this lack of standardization into account; in Figure 1, the top-left graph contains much implicit information, since unlabeled nodes represent carbons with valencies satisfied by hydrogens, and the top-right graph is fully labeled; that is, all atoms are explicitly labeled with their symbols. The former is what is usually exchanged informally between chemists, but actually mining chemical structures computationally requires something like the latter; for example, graph-mining algorithms can be applied to labeled graph representations to discover patterns in large-scale datasets,37 information that can also be determined through simulations (such as of molecular dynamics).
Though graphs implicitly encode the 3D structure of a molecule (when combined with knowledge about favored bond angles and distances), many different low-energy 3D structures, or conformers, may be consistent with the same graph structure. Moreover, the 3D structure may have a “secondary” geometrical arrangement of features (such as a right-handed helix) that cannot be encoded in the graph. Chemists thus have 3D representations (see Figure 1) that make explicit the 3D arrangement of atoms and bonds; the representation at the bottom right goes further, showing a molecular surface and some property that varies over the surface (such as lipophilicity, or the ability to dissolve in non-polar solvents). Even though some structure representations contain more explicit information than others, all are equally valid. When searching for substructures, a 2D representation suffices, but when exploring protein binding, a 3D structure is preferred; see the online Appendix for a notable subtlety in which the number and types of atoms and their connectivity may not uniquely define a structure.
A principle of cheminformatics is that similar molecules exhibit similar properties;16 the choice of representation is key in determining how such similarities are evaluated and thus the effectiveness of subsequent analyses. But there is a further challenge in balancing computational costs with the utility of a representation; for example, a full 3D description of a molecule accounting for all possible conformers would allow more accurate prediction of many properties, though such conformer predictions would also depend on good-quality force fields and algorithms. But the size of the representation and the time required for its evaluation would be prohibitive. So how can accurate similarity predictions be obtained from a subset of conformers? Or how can comparable accuracy by obtained through a 2D representation? Moreover, if it can be, what type of labels are required? Many such questions are answered today by trial and error through definition of an objective function (usually root mean square error or percentage correct) and iterative adaptation of descriptors and modeling approaches to optimize the objective function. No unifying theory is yet able to explain or even suggest optimal approaches in all cases.
A principle of cheminformatics is that similar molecules exhibit similar properties; the choice of representation is key in determining how such similarities are evaluated and thus the effectiveness of subsequent analyses.
Cheminformatics aims to support better chemical decision making by storing and integrating data in maintainable ways, providing open standards and tools allowing application and data use across heterogeneous platforms and mining the many chemical property spaces in a time- and space-efficient way. However, to flourish, cheminformatics needs closer collaboration between chemists and computer scientists, with the former able to pose their problems in a way that is relevant for practical applications and the latter to devise ways of capturing, storing, and analyzing chemical data to achieve optimal balance of space, performance, and complexity; for a detailed introduction to cheminformatics, written for computer scientists, see Brown.6 Cheminformatics is broadly divided into three areas: capturing data using lab notebooks or potentially using such formats as the Chemical Markup Language for publications; storing data (such as designing data base schemas and devising ontologies); and mining data (such as for predicting biological activity of compounds).
Bridging Cheminformatics and Computer Science
Here, we highlight current topics in cheminformatics, presenting computational and algorithmic problems and how computer science can contribute to their solution; for more, see the Appendix. We use as example cheminformatics methods “risk minimization” in drug discovery, minimizing the chances of a small molecule failing due to poor physical, chemical, or biological properties during research and development as a drug candidate; for example, a molecule must be soluble and show a certain degree of bioavailability to be considered a drug candidate. In cases where these properties are poor, a cheminformatics approach can suggest replacement of certain functional groups (connected sets of atoms affecting the characteristics of the chemical reactions of the molecule) to maintain potency but improve the solubility and bioavailability; see the Appendix for example properties a drug candidate must satisfy to be considered therapeutically useful and the role of cheminformatics at each stage of drug development.
Representing and searching structures. Most cheminformatics applications rely on large databases of chemical structures and their properties and relationships to, say, biological targets. Organizing and maintaining them, as well as searching and clustering similar structures together, are essential for many scientific applications. However, each of these areas poses computer-science challenges; for example, chemical and bioactivity databases (such as ChEMBLdb and PubChem) are freely available and contain possibly millions (ChEMBLdb) and tens of millions (PubChem) of data points. Integration of this disparate data is essential for researchers to gain the fullest possible perspective on what is presently known, tracking ongoing advances in science as they become available. Integration of data across chemical databases is a challenge due to sheer data volume and to the difficulties in normalization of chemical and bioactivity data. Molecular graphs must be captured in a machine-readable fashion. It is also necessary that life scientists be able to search chemical data (such as multilabeled graphs where graph labels can change from database to database). Just as labels can differ between databases chemical graphs can be encoded in multiple ways, depending how the nodes are ordered, resulting in multiple representations of the same molecule. The challenge for database search increases when considering protonation states (how the molecule changes when one proton is added to it) and tautomer states (how the molecule can change when a proton migrates from one part of the molecule to another). The need for a unique representation that is invariant with respect to atom ordering arises due to the expense of graph isomorphism (checking whether two structures represent the same molecule). Morgan23 described the first such “unique representation algorithm,” or canonicalization algorithm, allowing chemists to generate unique string representations of chemical graphs and compare structures through string comparisons. The Simplified Molecular-Input Line-Entry System, or SMILES, format defined by Weininger in 198839 is an example of a representation that can be canonicalized. Since the original canonicalization algorithm was proprietary, multiple implementations of the format have become available, each employing a different canonicalization algorithm, usually based on the Morgan algorithm; see Warr38 for an extensive discussion on chemical-structure representations.
Each database using its own algorithm for molecular encoding hinders the automated exchange of data between different databases. As more and more data has been made available online, the use of unique structure-based identifiers became a pressing need, resulting in development of the International Union of Pure and Applied Chemistry’s International Chemical Identifier (InChI), a non-proprietary, structured textual identifier for chemical entities.32 InChI identifiers are not intended to be read and understood by humans but are useful for computational matching of chemical entities. For quick database lookups, the InChIKey is a hashed key for the InChI with an invariant length of 14 characters; Figure 2 outlines the SMILES, InChI, and InChIKey for lipoic acid.
InChI is widely used for matching identical chemical structures but is still limited; for example, it cannot differentiate between certain types of stereoisomers (informally, molecules that are “3D mirror images” of one another) and is sensitive to tautomeric chemical forms; see the Appendix for a discussion of two stereoisomers for which the generated InChI is the same.
InChI and other identity-mapping algorithms allow for exact searching; two other practically relevant algorithms for scientific discovery based on chemical databases are substructure searching and similarity searching required to generalize from the search molecule to other, related molecules. In substructure searching, the database is searched for a specified wholly contained part of the search structure; in similarity searching, structures are retrieved that are similar (in some structure or property space) to the provided search structure. Chemical search packages are often implemented and optimized for a given database technology; for example, the OrChem package is an open-source chemical search package for the Oracle database application.27
For chemists it is important to appreciate that chemical space (occupied by all possible chemical structures) is, in principle, infinite.
Graph substructure matching is a variant of graph isomorphism, widely viewed as computationally intractable;8 executing a graph isomorphism search across a full chemical database of thousands or millions of structures is simply not feasible.40 Speedups can be obtained through structural fingerprint filters; fingerprints encode characteristic features of a given chemical structure, usually in a fixed-length bitmap. Fingerprints fall broadly into two categories: structure keys and hashed keys. In structure keys, each bit position corresponds to a distinct substructure (such as a functional group); examples are MACCS and PubChem keys. In hashed keys substructural patterns are represented as strings and then hashed to a random bit position; as a result, a given position can encode multiple substructures. The advantage of fingerprints is they can cover an arbitrarily large collection of substructures (such as “paths of length N,” or circular environments); examples are “daylight fingerprints” (“folded” to optimize information density and screening speed) and “extended connectivity fingerprints,” or ECFPs, that use local topological information. Given a binary fingerprint, chemists can first pre-screen a database to ignore molecules that cannot possibly match the query by requiring all bits in a query fingerprint to also be present in the target fingerprint. Since the target fingerprints are pre-computed, performing this check on modern hardware is quick. As a result, chemists apply the actual isomorphism test on only those molecules that pass the screen. Fingerprints can also be used to quickly search databases for similar molecules, using a similarity metric (such as the Tanimoto coefficient) to compare the query and target fingerprints. Additional heuristics34 further speed up similarity searches.
Molecules in their biological context. In the quest to discover novel therapeutic agents, studying molecules in a biological context is essential, as metabolites, cofactors, hormones, and signaling molecules. Developers of chemical databases must thus represent and organize chemical data across biological databases (such as those with pathways, protein information, and biological samples). Integration and processing data from such disparate domains underlie systems-level biological research. This yields additional challenges standard chemical structural representation cannot address: first, the representation of classes of chemical entities, since, in many cases, a class of compounds behaves in a certain context in a biological system rather than as a single molecule with a fully specified structure and, likewise, chemical concepts (such as groups, or parts of molecules); second, the need to represent non-structural groupings of compounds of interest, since compounds may bind to the same or similar targets or have similar biological functions (such as acting as an anti-neoplastic, preventing development of tumors). With multiple biological databases having to refer to these different chemical entities in an organized, standardized fashion to enable cross-database integration for a whole-system perspective, ontologies and other semantic technologies are used to support annotation, classification, and semantic cross-domain querying of chemical entities within biological systems. The most widely used ontology for biologically relevant chemical entities—ChEBI9—contained (as of February 2012) approximately 27,100 entities in an interlinked semantic graph and was used for annotation in dozens of biological databases. Ontologies are based on logical languages of varying levels of expressivity, accompanied by sophisticated reasoning algorithms. They can be used to construct various sorts of semantic similarity (such as in function and in application) that complement traditional structural-similarity methods.11 An open challenge is how to integrate graph-based chemical-structure information with wider logic-based ontology information to allow combined automated reasoning and querying over the different domains encoded in a growing number of interlinked ontologies. Enabling automated inference in such heterogeneous linked systems brings chemists closer to a systems-level understanding of small-molecule activity.26
Activity mining and prediction.
The basis of predictive modeling in cheminformatics is that the biological activity of a molecule is a function of the molecule’s chemical structure. Together with the similar-property principle16 mentioned earlier, the goal of any modeling approach is capturing and characterizing correlations between structural features and observed biological activity. Such approaches must also describe the likelihood of error when using the models for decision making. A variety of approaches can be employed to assess the error in (or conversely, the reliability or confidence of) a prediction, ranging from statistical approaches to more empirical approaches (such as defining an applicability domain, the region of input that can be predicted reliably, usually delineated by similarity to the training set).
In cases of receptor-mediated activity, the activity of a small molecule is due to its interaction with a receptor. Traditionally, quantitative structureactivity relationship (QSAR) model12,14 approaches do not consider receptor features, focusing instead on only small-molecule features, thus losing valuable information on ligand-receptor interactions. As a result, techniques (such as docking, which predicts how molecules fit together), structure-based pharmacophore modeling (a 3D approach to capturing protein-ligand interactions), and proteochemometric methods have been designed to account for both ligand and receptor structures. Proteochemometric methods are an extension of statistical QSAR methods to simultaneously model the receptor and ligand in a “system chemistry” sense, as first reported by Lapinsh et al.20
The first step in predicting biological activities is to generate molecular descriptors, or features, that are numerical representations of structural features; for example, labeled graphs and their associated characterizations are easily accessible to computer scientists yet miss significant physicochemical features (such as surface distributions and 3D pharmacophores). Chemists can also have difficulty objectively quantifying many chemical aspects of a molecule, such that the resultant descriptors are suitable for predictive modeling. Choosing a chemical descriptor should by no means be viewed as a “solved” problem; for more, see two comprehensive textbooks, one by Faulon and Bender,10 the other by Todeschini and Consonni.36
Molecular graphs can be transformed into numerical vector representations ranging from counts of elements to eigenvalues of the Laplacian matrix. Alternatively, molecular graphs can be compared directly through kernel methods, where a kernel on graphs G and G‘ provides a measure of how similar G is to G‘ or a kernel on a single graph compares measured similarities between the nodes of the two graphs. In cases where graph kernels have been defined, a chemist would, rather than compute vector representations, operate directly on the graph representations. Each method involves advantages and disadvantages; for example, the vector approach requires a computer scientist to identify a subset of relevant (to the property being modeled) descriptors; the feature selection problem is well covered in the data mining literature. A kernel approach does not require feature selection, but a computer scientist would face the problem of having to evaluate a dataset in a pairwise fashion, identifying an appropriate kernel. How to perform this evaluation is an important challenge, as the kernel must be selected to satisfy Mercer’s condition (a well-known mathematical property in machine learning that makes it easier to make predictions about a set of observations), and satisfying Mercer’s condition is not always possible with traditional cheminformatics-based kernels (such as those based on multiple common substructures). These challenges can make kernel-based methods computationally prohibitive on larger datasets.
Having settled on a numerical representation and a possible class of model types, a computer scientist would have to address the goal of the model. Is the chemist looking for pure predictive power, with no interest in explanatory features or more interest in decent predictive power, as well as some explanation as to why a molecule is predicted to be, say, toxic or nontoxic? The former scenario is common in virtual-screening settings, where the chemist might require a high degree of accuracy and fast predictions but not really care why one molecule is active and another inactive. Such models can be black boxes (such as neural networks) and algorithmic models5 (such as random forests). The latter is more common in exploratory and optimization settings, where a computer scientist might hope the output of the model will guide chemists in chemical modifications to improve the property being modeled. In this case chemists must understand the effect a certain structural feature has on observed potency, expecting the model to provide insight. Such models are generally distributional (such as linear regression, partial least squares, and naïve Bayes), though some algorithmic approaches can also provide insight.
Having chosen a modeling approach, the chemist and computer scientist must address model reliability closely tied to the concept of model applicability, or the reliability of the prediction of the model for a new object. This issue has become more important with increasing use of predictive models in regulatory settings. Misprediction (due to the model not being applicable to a certain class of inputs) can have significant financial repercussions. Various methods have been developed that attempt to characterize the model’s domain, determining not only whether models are applicable but whether additional biological experiments are required to reduce the prediction error on certain compound classes.
A key challenge faced in predictive modeling is the fact that small molecules are not static and do not exist in isolation. Though, traditionally, predictive models have focused on a single structure for a small molecule and ignore the receptor, small molecules can exist in multiple tautomeric forms and conformations. Enhancing the accuracy of predictions ideally requires taking into account the 3D geometries of the molecule and the receptor as much as possible. Though it is possible to generate reasonable low-energy conformations ab initio, the “biologically relevant” conformation might differ significantly (in terms of energetics) from the lowest-energy conformation of the molecule considered in isolation, necessitating conformational search. Multi-conformer modeling was addressed by the 4D-QSAR methodology described by Hopfinger et al.1 More recent techniques (such as multiple-instance learning) are also applied to the multi-conformer problem.
With the advent of high-throughput screening technologies, large libraries of compounds can now be screened against multiple targets in an efficient manner. Such panel assays provide a broad, systems-level view of small-molecule activities. Models developed on such data afford the opportunity to identify targets and characterize off-target effects. However, most approaches to the problem of screening multiple targets tend to develop multiple individual models,7 leading to multiple, independent predictions for a given input molecule. Alternatively, a system chemist (chemogenomics scientist) might imagine an approach that takes into account the covariance structure of multiple observed activities and structural descriptors within a single model. Such an approach could lead to more robust predictions for panel assays, or which battery of tests would be most useful. Finally, this approach also better reflects clinically relevant compound profiles19 and the “personalized medicine” concept (such as drug-drug interaction profiles).
Expanding chemical space. Enumerating molecules is a combinatorial problem that has fascinated chemists, computer scientists, and mathematicians alike for more than a century. Indeed, many fundamental principles of graph theory and combinatorics were developed by British mathematician Arthur Cayley, Hungarian mathematician George Pólya, and others in the context of counting isomers of paraffin. In the 1960s, Joshua Lederberg of Rockefeller University, Carl Djerassi of Stanford University, and others developed algorithms to enumerate structures based on spectral data, leading to DENDRAL, widely viewed as the first expert system.21
From a risk-reduction point of view, efficient methods for enumerating structures allow chemists to explore new regions of chemical space (such as to bypass patents), generate structures that exhibit desired properties, and identify molecules matching experimental data. For chemists it is important to appreciate that chemical space (occupied by all possible chemical structures) is, in principle, infinite. Even considering molecules for just 30 heavy atoms, the size of this space is on the order of 1060 heavy atoms.4 Any enumeration method would face a combinatorial explosion if implemented naïvely.
A key application of structure enumeration is the elucidation of structures based on spectral data,17 especially for identifying metabolites, or small molecules that are the by-products of metabolic processes and thus provide insight into an organism’s biological state (such as diseased and fasting). A chemist gathers spectral data (such as nuclear magnetic resonance, mass, and liquid chromatographymass spectrometry), and an algorithm would ideally provide a list of structures that give rise to the observed spectra. Some commercial products (such as MOLGEN http://molgen.de) are able to perform this task quickly.
Another application of structure enumeration concerns Markush structures and searches;2 for example, a Markush claim used in a patent would cite multiple “functionally equivalent” chemical entities, where the user specifies a pattern (such as “…an aromatic ring with two alkyl groups attached to it”). Such a pattern is very general: an alkyl group can be only methyl, ethyl, or any chain of n carbons, with three possible positions for them on the ring. However, even this simple definition involves 3n2 possible structures. More complex Markushes can involve billions of possible structures. Explicit enumeration is not feasible. Analysis of Markush structures thus faces a number of challenges, primarily the ability to search (based on structural or property similarity) through the implicit enumerated space. A number of commercial vendors, including Digital Chemistry and ChemAxon, offer toolkits for these problems.
Given that any experiment could become the core of a patent, ELNs allow organizations to efficiently define and implement audit trails.
Structure enumeration plays a fundamental role in molecular design, or the design of compounds that optimize some physical, chemical, or biological property or activity.30 A key challenge is how to combine enumeration algorithms with efficient property prediction and is closely related to methods in predictive modeling of chemical properties discussed earlier. This approach is also termed inverse QSAR, where the chemist must devise a set of features that describe a molecule with a specified property and subsequently reconstruct the molecule from those features. Moreover, the reconstruction of molecules from feature sets can be viewed as a cryptography problem. Such reconstruction is relevant to the pharmaceutical industry, as it may be necessary to share data on molecules without also sharing explicit structures. Feature sets that allow easy reconstruction of molecular structures are thus undesirable. A number of chemistry researchers have investigated the problem of identifying such one-way molecular descriptors, as well as methods for reconstructing molecules from descriptors.22 New methods that generate such one-way molecular features but still correlate with physicochemical properties would be valuable.
While molecular-graph enumeration is a challenge, an alternative to enumerating molecular structures based on spectral data is to sample these structures.13 Sampling procedures based on metropolis or genetic algorithms help elucidate compounds from NMR data. However, even in the face of the complexity of molecular enumeration, computational chemists have developed and successfully used enumeration tools to generate large chemical libraries (such as GDB-13 http://www.cbligand.org/gdb13/) of almost one billion chemical structures3 considering only molecules up to 13 heavy atoms and using only carbon, nitrogen, oxygen, sulphur, and chlorine. However, current enumeration software products do not generally produce stereoisomers or tautomers that require specific enumeration procedures and are thus still the subject of cheminformatics research.
Another relevant application of enumeration methods is how to generate chemical-reaction networks. The problem here consists of enumerating all possible compounds that can be produced by applying reaction rules to a set of initial molecules. By reversing the reaction rules chemists are also able to find sets of starting compounds necessary for producing a given target, a process called retrosynthesis. Designing new drugs and chemicals, understanding the kinetics of combustion and petroleum refining, studying the dynamics of metabolic networks, and applying metabolic engineering and synthetic biology to produce heterologous compounds (compounds from different species) in microorganisms all involve enumeration of reaction networks. As reviewed in Faulon and Bender10 several network-enumeration techniques have been developed but generally suffer from a combinatorial explosion of product compounds. One way to limit the number of compounds being generated is to simulate the dynamics of the network while it is being constructed and remove compounds of low concentration.
Following this idea, methods have been developed based on the Gillespie Stochastic Simulation Algorithm (http://en.wikipedia.org/wiki/Gillespie_algorithm) to compute on-the-fly species concentrations. Chemical-reaction-network enumeration and sampling is an active field of research, particularly in the context of metabolism, for studying biodegradation or proposing metabolic-engineering strategies to biosynthesize compounds of commercial interest. However, a difficulty with metabolic network design is that, in addition to network generation based on reactions, a system chemist or biologist must also verify there are possible enzymatic events to enable reaction catalysis; enzymes must be present to reduce the energy required for some reactions to take place. This additional task requires computer scientists include both chemical structures and protein sequences and develop tools at the interface between cheminformatics and bioinformatics.
Knowledge management. Scientific-knowledge management is increasingly relevant not only to reduce risk in current research but to enable new collaboration and innovation opportunities with internal partners and a growing number of external (public) partners. In cheminformatics and chemistry, scientists switched years ago from paper lab notebooks to online collaboration platforms called electronic lab notebooks, or ELNs. So, what finally drove chemists and pharmaceutical companies to adopt “enterprise 2.0” social online-collaboration culture? Before 2000, many chemists still used paper lab notebooks and multiple compound-registration and search tools. The overall architecture was too inflexible to adapt to fast-changing data standards, and scientists spent too much time on administrative work; moreover, chemical data quality was inconsistent, and alignment with other working groups was inefficient (such as for running analytical experiments). Legal disputes would require manual searches through many paper lab notebooks, and data synchronization was painful, hindering large-scale collaboration.
While ELNs were available as long ago as the 1990s, chemists only began adopting them in large numbers in 2004 and 2005, and the market continues to grow.35 A 2006 report (http://www.atriumresearch.com/library/Taylor_Electronic_laboratory_notebooks.pdf) said, “The initial drive for the development of ELNs came from the field of chemistry, perhaps driven by the early adoption by chemists of computer technologies […] The pharmaceutical industry shifted from a position of ‘if we get an ELN’ to ‘when we get an ELN’.”35 The growth of ELNs is driven by many factors, including ease of use (rapid search across organizationwide data sources and easy sharing of domain knowledge) and regulatory compliance (time-stamped experimental data, experiment and data approvals, and quality-control information). Given that any experiment could become the core of a patent, ELNs allow organizations to efficiently define and implement audit trails.
“Electronic lab notebooks have replaced the traditional paper lab book across the pharmaceutical industry”18 but are not yet common in academia due to their cost. Worth mentioning here is that chemistry is traditionally a field accustomed to bookshelves laden with reactions and compounds. Chemists have switched to online ELNs not only to increase efficiency but because they allow easy collaboration with trusted colleagues. Third-party paper catalogues and academic publications proved inefficient and, critically, did not account for compounds from trusted colleagues. The change of management to ELNs, as with enterprise 2.0, has required delivering on the promise of greater interconnectivity and collaboration opportunities, made legally possible in the U.S. due to the U.S. Food and Drug Administration’s regulation Title 21 CFR Part 11, permitting electronic signatures in chemical documents. Leading players include CambridgeSoft’s E-Notebook and Accelrys’s Symyx] Notebook, but at least 35 different companies were producing ELNs as of February 201128 for chemistry, biology, quality control, and other research domains. Another knowledge-management tool designed specifically for chemists is the Web-based service Reaxys (https://www.reaxys.com/info/) launched in 2009 to provide “a single, fully integrated chemical workflow solution.”
However, using external (public) databases with chemical and bioactivity data is a challenge due to differences in identifiers, synchronization, curation, and error-correcting mechanisms, as well as the need to provide efficient substructure and similarity search within complex data types. A collaboration with external parties (such as contract research) poses further challenges, including compound duplication and efficient data synchronization. Some external partners may lack sufficient IT resources themselves and thus rely on external services. Cloud services could help provide a service infrastructure for all parties involved, as well as the required private-public interface. Furthermore, many data sources are still not being indexed properly; for example, chemistry patents are often cryptic, and chemical image and text mining is a challenge though is being addressed in academic and industrial research.15,29 The closed Chemical Abstract Service (CAS http://cas.org) is one highly trusted source, and public chemical and bioactivity databases must improve their quality and interconnectivity to compete. SciFinder (http://www.cas.org/products/scifinder/) is another chemical-abstract service, with a version, SciFinder Scholar, marketed to universities. Open-source efforts like ChEMBL and PubChem-Bio-Assay are on the right track, though, unlike some commercial tools, do not abstract reactions. Still, improving data quality and standards between public and closed sources is critical for ensuring growth, use, and collaboration by private and public parties alike.
Novel Algorithms
Essential for researchers interested in new algorithms is a software library that handles chemical structures and related data; a variety of such chemical toolkits are available, both proprietary (possibly involving free academic licenses) and open source.
Due to the importance of cheminformatics to the pharmaceutical industry, numerous commercial vendors provide libraries, applications, and database cartridges, including Accelrys, BioSolveIT, Chem-Axon, Chemical Computing Group, Daylight, Open Eye, Schrodinger, Tripos, and Xemistry. Since about 1995, open-source cheminformatics software has emerged, providing opportunities for rapid development and implementation of novel algorithms that build on the existing open-source ecosystem. Here, we do not debate open source, focusing instead on open-source tools to help explore cheminformatics problems.
Open-source software in cheminformatics has lagged open-source software in bioinformatics. However, recent years have seen a rise in open-source cheminformatics software. One notable organization is the Blue Obelisk group25 (http://blueobelisk.org) of chemists seeking to create interoperable open-source chemistry software and open-source chemical toolkits, including the Chemistry Development Kit (CDK),33 Open Babel,24 RDKit, and Indigo, written in Java or C++, though those in C++ have, via http//:swig.org, bindings to a variety of languages; for example, such toolkits are used to read/write chemical files in various formats, manipulate chemical structures, measure similarity of molecules, search for substructures, and generate 2D depictions. The underlying algorithms include graph algorithms (such as maximal common substructure and canonical labelling of graphs), geometrical methods (such as Kabsch alignment), and vector manipulation (such as converting coordinates in various systems and 3D structure generation). Besides being subject to programmatic use by cheminformaticians, many applications rely on these toolkits to handle chemical data; for example, the molecular viewer Avogadro (http://avogadro.openmolecules.net) uses Open Babel, and the molecular workbench Bioclipse31 uses CDK.
Though the toolkits share many features they also have certain distinguishing features; for example, CDK implements a large collection of molecular descriptors. Open Babel has robust support for 3D structure generation and optimization and supports the interconversion, or conversion of one file format to another, of a large number of chemical file formats; the Appendix includes a comparison of features offered by the open-source toolkits. Given there is ample scope for software engineering (such as algorithm implementation, code quality and analysis, and building systems), CDK and Open Babel are both open to new contributions, driven by public mailing lists and tracking systems.
Certain challenges faced by nearly all cheminformatics toolkits stem from the graph representation of chemicals, that, while succinct and amenable to computation, only approximates reality with edge cases abounding. Areas of interest include enumeration of colored graphs accounting for symmetry and symmetry detection itself, which not only derives from the chemical graph but typically also from the 3D geometry of the chemical. Chemists must realize, though the sophistication of chemical representations has increased over the years, many chiral molecules and non-standard bonding cases (such as organometallic compounds) cannot be handled by current representation systems. A further challenge is the chemical representation of a molecule would need to capture non-static graphs to account for delocalization and the tautomeric phenomena molecules undergo in real biological contexts.
Of equal importance to development of new algorithms is freely available data on which to train and test new methods. As noted earlier, large structure and bioactivity data collections are available, enabling much more robust validation of cheminformatics methodologies, as well as large-scale benchmarking of algorithms. Benchmarking is especially relevant for the data-mining techniques in cheminformatics. Open data focuses primarily on structure and activity data types, with a notable lack of “open” textual data (such as journal articles). While PubMed abstracts (http://www.ncbi.nlm.nih.gov/pubmed/) are a proxy for journal articles, text-mining methods in cheminformatics are hindered by not being able to mine the full text of many scientific publications. Patent information is publicly accessible, supporting these efforts. Open data does not explicitly address data quality or problems integrating data sources. In many cases, manual curation is the only option for maintaining high-quality databases; fortunately, open-data facilitates curation by allowing independent access to anyone with a computer.
Conclusion
Cheminformatics, the computer science of chemical discovery, is an industrial and academic discipline with roots dating to the 19th century and a flowering in the 1960s along with modern computing technologies. While many key cheminformatics techniques have been available in the literature since the 1960s, most cheminformatics tools and implementations have been proprietary; likewise, most data sources have been proprietary (usually with restrictive licensing policies) until recently. Companies look to protect their intellectual property for many reasons primarily involving profit, competitive intelligence, and intellectual property relevant to the pharmaceutical industry, as well as to other chemical-related industries. Unlike much of bioinformatics, these issues are where the data and tools have been freely available since the field’s inception. The disparity between cheminformatics and bioinformatics can be attributed to the fact that the outcomes from cheminformatics methods and software have a more direct effect on profits, in terms of identifying lead-like compounds and improving the properties of drug candidates, while bioinformatics software is found in upstream areas (such as target identification) and is perhaps less directly related to possible profits as a candidate small molecule. Moreover, acquiring chemical data (such as structure and activity) is more difficult and when done on a large scale can involve much time and effort, whereas acquiring bioinformatics data (such as sequences) is much easier.
Keeping in mind cheminformatics is fundamentally a practical field, serving experimental chemistry, the key challenges require an understanding by scientists of the underlying chemical systems.
While both fields have theoretical components, it is possible that the free availability of a large amount of bioinformatics data drove development of related publicly available tools. In contrast, the proprietary nature of chemical data would imply that tools needed to process and analyze it are primarily of interest to the owners. As a result, there has been little incentive to make cheminformatics software publicly available, and since users are primarily industrial, commercial software is the norm.
However, the cheminformatics landscape is shifting, with free open-source cheminformatics toolkits, applications, and open databases with tens of millions of compounds and experimental bioactivity data, while modern experimental techniques (such as high-throughput sequencing) have made generation of large amounts of structure-activity data much more accessible.
Prospects for academic and industrial collaboration between chemists and computer scientists are bright, encouraging more computer scientists to participate in cheminformatics research; for many specific open research questions, see the Appendix.
Significant domain knowledge is required to address problems in cheminformatics. Indeed, many issues faced by chemists are not as clean an abstraction as “algorithms on a string,” (substring matching is not very useful on multilabeled graphs) the way many bioinformatics algorithms can be abstracted into a computer-science framework. Hence, while chemistry researchers can contribute to cheminformatics simply by considering structures as graphs, they are inevitably limited to somewhat abstract problems. Keeping in mind cheminformatics is fundamentally a practical field, serving experimental chemistry, the key challenges require an understanding by scientists of the underlying chemical systems.
Nevertheless, due to increasing availability of tools and data, the barrier to entry for non-chemists and non-cheminformaticians is significantly lower than a decade ago. Many questions that would benefit from computer science can now be addressed; for example, for theorists, graph-theoretic questions of 3D enumeration; for database designers, more effective search algorithms and ontologies; for practical programmers, expansion of the many open-source cheminformatics projects. If more chemists would think algorithmically and more computer scientists chemically, the pharmaceutical industry and associated industries would be much better positioned to deal with not only “simple” chemicals (only one identifier and isomer possible per molecule) but also their complex relationships, transformations, and combinatorial challenges, bringing cheminformatics closer to the goal of supporting primary translational research in the wider context of chemical biology and system chemistry.
Acknowledgments
We would like to thank Danny Verbinnen of Janssen Pharmaceutical Companies for sharing his insight into ELNs and John Van Drie of Van Drie Consulting for his insight into cheminformatics. This article was written while Aaron Sterling was visiting the Department of Electrical Engineering and Computer Science at Northwestern University where he was supported in part by National Science Foundation grant CCF-1049899. Andreas Bender thanks Unilever for funding. Janna Hastings thanks the European Union project EU-OPEN-SCREEN for funding.





{kind=link}
{kind=link}
Join the Discussion (0)
Become a Member or Sign In to Post a Comment